
壞死性硬化是程序性細胞死亡的一種裂解形式,與促炎性細胞因子的產生,生物膜的破壞以及細胞內損傷相關分子模式(DAMPs)的釋放有關。壞死的依賴於混合譜系的激活激酶結構域樣(MLKL)假性通過受體相互作用蛋白激酶3(RIPK3)。MLKL的RIPK3介導的磷酸化觸發的構象變化它促進了向細胞膜的移位和最終不可逆的破壞。而膜破裂的精確生物物理機制仍然是有爭議的問題,現代的機型的共同特點是一個MLKL低聚物的形成和劊子手四螺旋束結構域的直接關聯MLKL的(4HB)與生物膜。
我們已經確定了小鼠Mlk1的單個鹼基對種系突變,該突變編碼MLKL括號區域內的錯義取代,並賦予獨立於上游壞死性刺激的組成性激活。鑑於此突變Mlk1等位基因與野生型Mlk1一樣受到基因表達的發育和環境控制,這些小鼠的出生後致死率使壞死性壞死病的生理和病理後果更為深刻。同時,這些發現揭示了三種常見的人類MLKL多態性的潛在功能意義,這些多態性編碼非保守性氨基酸取代,位於突變的大括號螺旋內或附近。
MLKL組成型活性形式的產生
由於血小板生成素主要受體的基因缺失,MPI -/-小鼠的外周血血小板數量只有野生型的10%。進行了ENU誘變篩選,以鑑定通過血小板生成素非依賴性血小板生成改善Mpl -/-小鼠中血小板減少的突變。AG 1的創建者,稱為Plt15,與Mpl -/-動物的平均值(113±57×10 6 / mL)相比,血小板計數適度升高,為189×10 6 / mL ,產生了19 Mpl -/-後代。這些小鼠中有十隻的血小板計數超過200×10 6每毫升,與主要作用突變的分離一致(圖 1a)。連鎖分析和測序鑑定出在Mlk1中A到T的轉位在所有血小板計數升高的小鼠中都是雜合的(圖 1b)。
所述Mlkl Plt15突變導致非保守天冬氨酸到纈氨酸置換在所述第一支撐螺旋內的位置139。在全長mMLKL結構中,D139形成一個鹽橋,在MLKL四螺旋束(4HB)域0位(α2螺旋)處帶有一個精氨酸殘基(圖 1c)。此鹽橋代表MLKL 4HB結構域螺旋α2中的殘基與兩螺旋“括號”區域之間的一系列靜電相互作用之一。迄今報導,小鼠MLKL的D139在椎骨的所有MLKL直系同源物中均是保守的(圖 1d)。
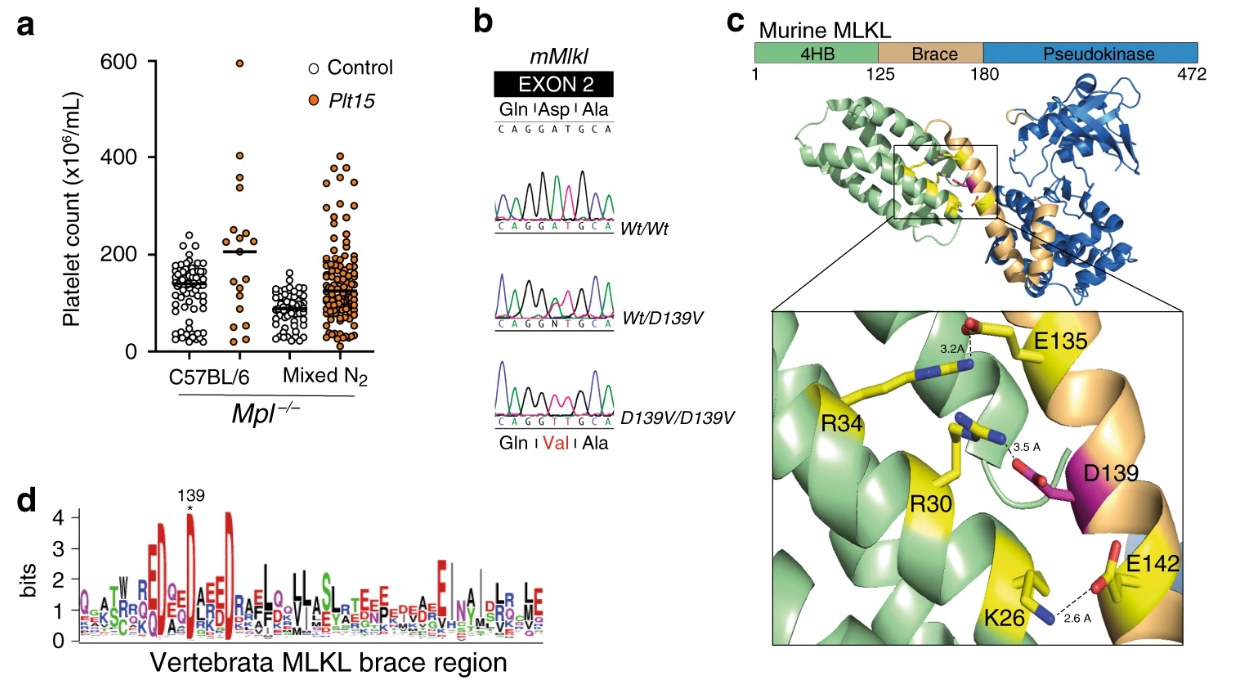
MLKLD139V老鼠是MLKL的組成型活性形式。(圖源:Nature)
常見的人類錯義MLKL變體映射到支撐區域
鑑於鼠Mlk1 D139V / D139V新生兒的嚴重炎症表型和在鼠Mlkl Wt / D139V成人中觀察到的造血功能嚴重缺陷,我們探討了人MLKL支架區域變異的普遍性。檢驗gnomAD數據庫,其中包含來自總共140,000多個個體的人MLKL外顯子組或基因組序列數據,顯示第二和第三高頻率的人MLKL錯義編碼變體。rs34515646(R146Q)和rs35589326(S132P),更改相同的括號螺旋。第四大人類MLKL多態性,rs144526386(G202 * V)是只在MLKL(*)命名為’MLKL2’的較短同種型剪接的上下文識別的錯義多態性。MLKL的全長標準轉錄本編碼一個471個氨基酸的蛋白質,而MLKL2是MLKL的另一個剪接同工型,長度為263個氨基酸。MLKL2缺乏假性域的大部分要抑制上述4HB域的殺傷潛力發揮功能和招共效應等RIPK3和HSP90。甘氨酸202 *由外顯子9的延伸編碼,這是MLKL2剪接同工型所特有的(圖 5a,b)。
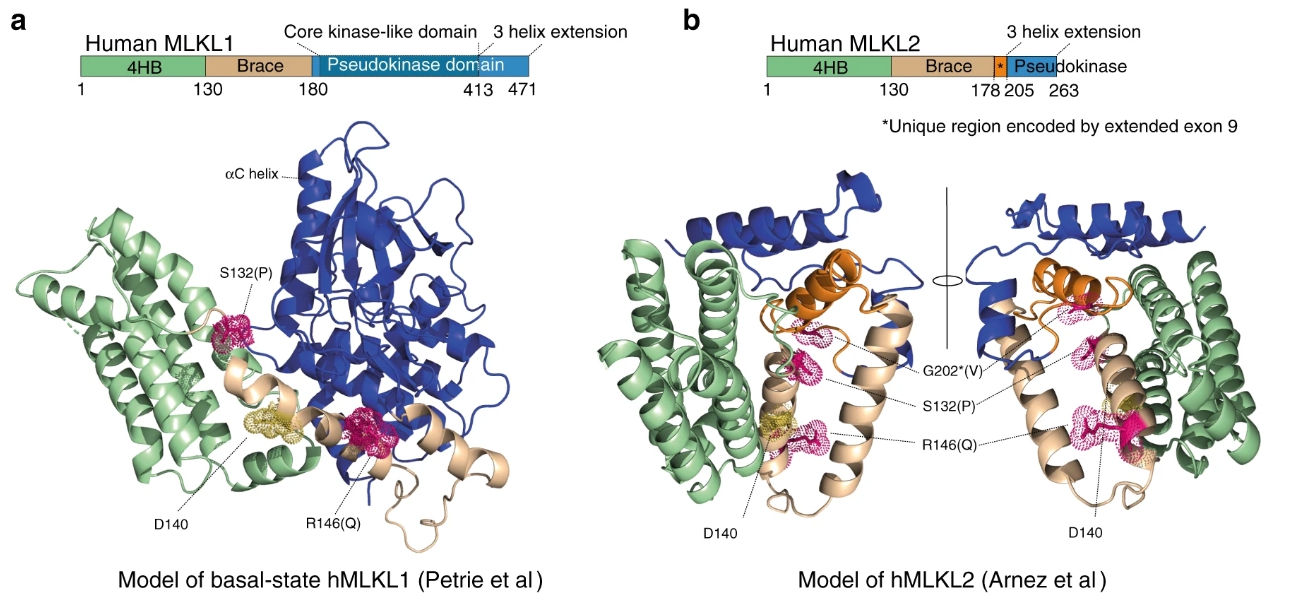
四個最高頻率錯義人MLKL SNP中的三個編碼括號螺旋區域內或附近的非保守氨基酸取代。(圖源:Nature)
Mlkl D139V導致致命的圍產期炎症綜合症
為了定義組成型活性MLKL在沒有Mpl缺乏導致的任何混雜效應的表型後果的情況下,所有後續研究均在Mpl + / +背景下進行。純合子Mlkl D139V / D139V幼犬出生於預期的孟德爾頻率,表面上在肉眼和組織學上在E19.5看來正常。然而,到三天大時,儘管它們與同窩幼仔在外觀上沒有區別(圖 2a),但它們的體重卻有所減輕並沒有壯成長,在常規清潔住房條件下的最長觀察壽命為6天。像Mlk1 Wt / D139V小鼠一樣,Mlk1 null / D139V複合雜合子在P21處以預期的頻率出現,並正常發育至成年。
因此,MLKL D139V的組成性活性不受正常MLKL蛋白的存在的影響,表明決定圍產期致死率的是Mlk1 D139V的絕對等位基因劑量。為了證實源自ENU的Mlk1 D139V小鼠的表型是由於Mlk1 D139V引起的錯義突變,我們使用CRISPR-Cas9基因組編輯獨立生成了Mlk1 D139V小鼠。純合子CRISPR- Mlk1 D139V / D139V小鼠出生後也很快死亡。
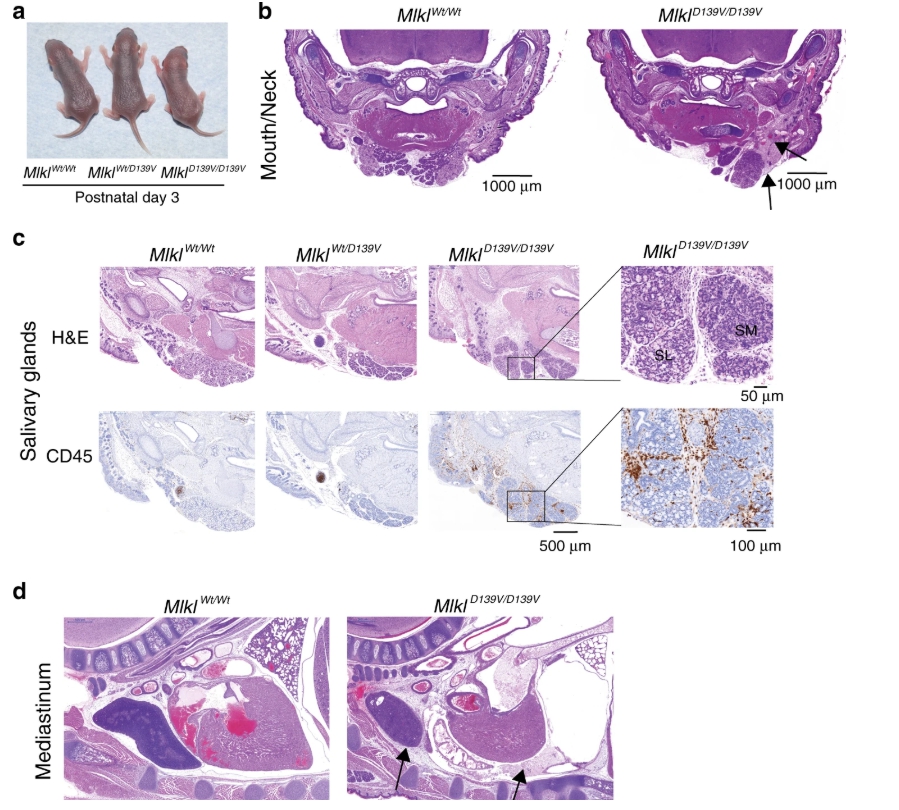
純合Mlk1 D139V新生兒表現出分散的上身炎症。(圖源:Nature)
Mlkl D139V小鼠的造血缺陷
儘管相對於Mlkl Wt / Wt和Mlkl Wt / D139V同窩仔,E19.5的Mlkl D139V / D139V幼犬的血細胞數量沒有變化,但通過P3,白細胞總數明顯不足(主要是由於淋巴細胞數量減少)和血小板數量(圖3a–c)。類似地,儘管活細胞中細胞內ROS的增加水平一致,但在E18.5 Mlk1 D139V / D139V幼仔的胎兒肝臟中,造血幹細胞和祖細胞的數量以正常比例存在(圖 3d,e)。通過P2,在CD150赤字+ CD48 +和CD150 + CD48 -群體存在(圖 3f),伴隨著增加膜聯蛋白V在活細胞的結合(表示磷脂酰絲氨酸曝光)所有譜系(圖的 3g)。在成年Mlkl Wt / D139V小鼠中,造血幹細胞和祖細胞的數量不受影響(圖 3h)。
當受到細胞毒性藥物5-氟尿嘧啶(5-FU)的攻擊時,Mlkl Wt / D139V小鼠同樣受到延遲(圖 3i)。在競爭性移植中,如預期的那樣,將Mlkl Wt / D139V或Mlkl Wt / Wt骨髓與野生型競爭者骨髓一起以10:1的比例注射,Mlkl Wt / Wt骨髓在8週內貢獻了90%的受體血細胞移植後維持該貢獻水平達6個月(圖3j)。相比之下,Mlkl Wt / D139V骨髓表現較差,在這些時間貢獻了25%和51%的受體血細胞(圖 3j)。 )。類似地,儘管野生型胎兒肝細胞在移植後直至6個月內在受輻照的受體中佔絕大多數血細胞,但在此期間,來自Mlk1 D139V / D139V胚胎的細胞未能有效競爭(圖3k )。雜合子Mlkl Wt / D139V胎兒肝細胞在移植後的第一個月中貢獻較弱,但在六個月後恢復以貢獻更多(圖 3k)。因此,儘管在穩態條件下可耐受,但Mlk1 D139V的雜合性在造血應激條件下是有害的。來自Mlkl Wt / D139V的骨髓源性HSC成年小鼠和來自Mlkl Wt / D139V和Mlkl D139V / D139V幼崽的胎兒肝源性造血幹細胞在8天后,在經致死劑量照射的受體小鼠的脾臟中也形成了越來越少的集落。
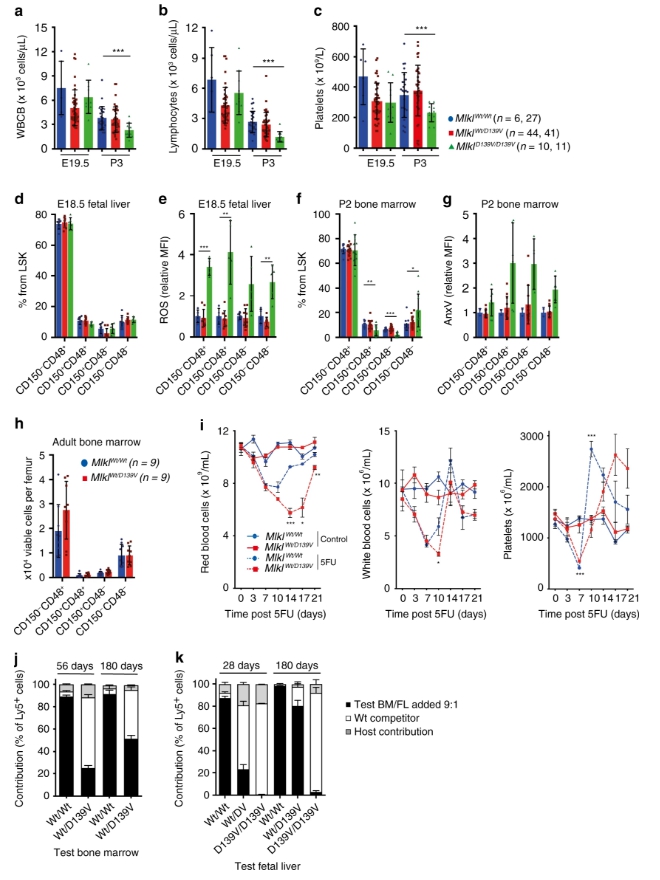
Mlk1 D139V小鼠的造血細胞改變和有缺陷的緊急造血功能。(圖源:Nature)
MLKL括號變體在CRMO中更頻繁地出現
為了研究人MLKL支架區域多態性是否在人自身炎性疾病中起作用,我們檢查了其在強直性脊柱炎(AS),慢性複發性多灶性骨髓炎(CRMO),格林巴利綜合徵(GBS)和滑膜炎,痤瘡,膿皰病,骨肥大和骨炎(SAPHO)綜合徵。當考慮人群分佈時,相對於健康對照組,R146Q,S132P和G * 202V的個體次要等位基因頻率並未豐富。但是,這些等位基因發生在反式。這是在健康的NIH 1000基因組樣本中觀察到這些組合的頻率的29倍,或者是只有歐洲CRMO患者和比較了兩個獨立的歐洲健康對照人群。
與細胞凋亡相反,壞死病被廣泛認為是細胞死亡的一種炎症形式。但是,有關這一主張的確切證據尚未出現。由於MLKL被諸如TNF的炎性刺激所激活,因此很難將原因與效果分開。小鼠中MLKL(Mlk1 D139V)的自激活突變體的偶然鑑定使我們能夠探索在沒有此類混雜因素的情況下不適當的屍檢的後果。此外,它對重要的成年造血和圍產期發育過程(對過度的MLKL活化最敏感)以及導致中和活化的MLKL的生理機制產生了深刻的見解。
試圖以結構,生化,細胞和動物為基礎的功能證據覆蓋,試圖推測這些人MLKL支架區域變異導致MLKL功能和/或調節發生改變,這很可能是高度組織,背景或什至是病原體特異性方式。雖然將需要更多的人數和獨立隊列的檢查來確認自發炎性疾病CRMO 中反式發生的人MLKL支架變異的統計富集,但該患者隊列為他們目前作為複雜的多基因炎症性疾病的調節劑提供了誘人的線索天人類。
參考資料: